Диетотерапия при фенилкетонурии
Содержание:
Примерное меню новорожденного с ФКУ 3-х недельного возраста с массой тела 3500гр Толерантность фенилаланина 140 мг/сутки
| Кол-во (мл) | Фенилаланин (мг) | Белок (г) | Жиры (г) | Углеводы (г) | Энергия (ккал) | |
|---|---|---|---|---|---|---|
| Диетическая норма на кг/сутки | 40 | 2,4 | 4,9 | 13,6 | 108 | |
| Диетическая норма в сутки | 140 | 8,4 | 17,15 | 47,6 | 378 | |
| Грудное молоко PKU 1- mix (49г + 293 мл воды) = | 312 | 140 | 3.43 | 12.48 | 22.15 | 221.52 |
| 323 | 4.96 | 13.52 | 27.58 | 251.86 | ||
| Суточная норма | 140 | 8,39 | 26 | 49,73 | 473,38 |
Ребёнок может получить
- 3 x грудное молоко в объёме 104 мл и 4 x Milupa PKU 1-mix 94мл (на 1 порцию 12,2г порошка или 2,8 мерки) или
- 7 x 54мл Milupa PKU 1-mix (на 1 порцию 7,7г препарата или 1,8 мерки) и 7 x 44 мл грудного молока
| Кол-во (мл) | Фенилаланин (мг) | Белок (г) | Жиры (г) | Углеводы (г) | Энергия (ккал) | |
|---|---|---|---|---|---|---|
| Диетическая норма на кг/сутки | 40 | 2,4 | 4,9 | 13,6 | 108 | |
| Диетическая норма в сутки | 140 | 8,4 | 17,15 | 47,6 | 378 | |
| Грудное молокоXP Analog LCP (38г+228 мл воды) = | 312 | 140 | 3.43 | 12.48 | 22.15 | 221.52 |
| 252 | 4.94 | 8.74 | 20.52 | 180.5 | ||
| Суточная норма | 140 | 8,37 | 21,22 | 42,67 | 402,02 |
Ребёнок может получить
- 3 x грудное молоко в объёме 104 мл и 4 x 63мл XP Analog LCP (на 1 порцию 9,5г порошка или 1,9 мерки) или
- 7 x 36мл XP Analog LCP (на 1 порцию 5,0г препарата или 1,0 мерка) и 7 x 45мл грудного молока
Статистика фенилкетонурии в Польше
В Поликлинике Метаболических Заболеваний ИМиР (Польша) наблюдается
-
* В общем – ok. 780 больных
-
— мягкая гиперфенилаланинемия – 172 человек
-
— классическая фенилаланинемия — 595 человек
-
— злокачественная гиперфенилаланинемия —
-
дефицит BH4 – 12 человек
Дети с ГФА, рожденные в 2010 году:
-
В общем – 26 новорождённых
-
(полная диагностика: общие анализы крови, фен/тир; двугидроптериновая редуктаза DHPR, тест нагрузки BH4, профиль выделенных из мочи биоптеринов)
-
— 10 детей – классичеслая ФКУ
-
— 1 ребёно — мягкая ФКУ
-
— 12 детей – мягкая ХФА
-
— 1 ребёнок – дефицит PTPS
-
— 2 детей — переходная гиперфенилаланинемия
«Потерянные» случаи — причины
-
Рождённые дома
-
Ранняя выписка из больницы
-
Ошибка в подписи пробы (один код на пробе, другой код приклеен в детской книжке)
-
Забыли взять пробу крови
-
Потерия пробы крови по пути в лабораторию
-
Проба крови взята от одного ребёнка на несколько бумаг
Рекомендации
-
Необходимость проведения скрининга всей популяции новорожденных.
-
Проведение диагностики разными методами в каждом случае гиперфенилаланинемии.
-
Начало диеты с низким содержанием фенилаланина как можно раньше, оптимально до исполнения 21 суток жизни ребенка.
-
Начало лечения сразу при уровнях фенилаланина > 10 мг %, или после нескольких дней ежедневного контроля, если концентрация фенилаланина удерживается в пределах 7-10 мг %
-
Проведение лечения на основе современных препаратов без фенилаланина согласно определенным схемам, с учетом в каждом случае индивидуального потребления фенилаланина.
-
Регулярный мониторинг избранных биохимических показателей в крови больного (например, концентрации фенилаланина, тирозина, цельного белка) а также психического и физического развития больных.
-
Лечение девушек и женщин до зачатия, а также в течение всего периода зачатия, из-за вредного действия высокой концентрации фенилаланина на развитие плода.
-
Продолжение лечения и мониторинг лечения больных взрослых.
Недостаток однозначных лечебных рекомендаций для взрослых
-
„Исчезание” пациентов после окончания 18 года жизни
-
Проблема материнской фенилкетонурии
-
Проблема остеопороза при длительной диете
-
Низкий уровень акцептации („compliance”) у подростков и взрослых обусловлен чрезвычайной обременительностью лечения
-
(правильно лечится меньше, чем 20% взрослых больных)
Механизм развития и причины заболевания
Причина возникновения этого заболевания связана с тем, что в печени человека не вырабатывается особый фермент – фенилаланин-4-гидроксилаза. Он отвечает за превращение фенилаланина в тирозин. Последний входит в состав пигмента меланина, ферментов, гормонов и необходим для нормальной работы организма.
При ФКУ фенилаланин, в результате побочных путей обмена, превращается в вещества, которых не должно быть в организме: фенилпировиноградную и фенилмолочную кислоты, фенилэтиламин и ортофенилацетат. Эти соединения накапливаются в крови и оказывают комплексное действие:
- нарушают процессы жирового обмена в мозге;
- оказывают токсическое действие, отравляя мозг;
- вызывают дефицит нейромедиаторов, которые передают нервный импульс между клетками нервной системы.
Это вызывает значительное и необратимое снижение интеллекта. У ребенка быстро развивается умственная отсталость – олигофрения.
Заболевание наследуется только в том случае, если оба родителя передали ребенку склонность к болезни, и поэтому встречается довольно редко. У двух процентов людей есть измененный ген, который отвечает за развитие болезни. При этом человек остается полностью здоровым. Но когда мужчина и женщина, носители мутировавшего гена, вступают в брак и решают завести детей, то вероятность того, что малыши будут страдать от фенилкетонурии, составляет 25%. А возможность того, что дети будут носителями патологического гена ФКУ, но сами останутся практически здоровыми, составляет 50%.
Причины в наследовании
Как и при других ферментопатиях, наследование происходит по аутосомно-рецессивному типу. Данный тип наследования указывает на наличие в аутосомных хромосомах рецессивного гена, проявление заболевания возможно лишь в гомозиготном состоянии.

Другими словами, у родителей, не страдающих фенилкетонурией, но являющихся носителями признака, вероятность рождения больного ребенка составляет 25%, при одном больном родителе – 50%, если оба родители больны – стопроцентная вероятность рождения ребенка с данной патологией. Примерная частота 1 случай на 10-11 тысяч новорожденных. При этом часто при сборе генетического анамнеза выявляется кровное родство родителей (примерно каждый 10 случай).
Симптомы фенилкетонурии
Симптомы заболевания у детей с классической формой патологии сразу после рождения не наблюдается. Однако такие дети обладают рядом специфических внешних признаков. У них имеет место быть:
- суховатая кожа белого оттенка;
- пигментация отсутствует практически полностью;
- волосы светлого оттенка;
- голубые глаза.
В возрасте 2 — 6 месяцев начинают проявляться симптомы заболевания. К вышеуказанным признакам добавляется вялость, пассивное восприятие окружающего мира, повышенная раздражительность и задержка психомоторного развития. Иногда возможно возникновение частой рвоты. Появляется беспричинное беспокойство. Могут наблюдаться приступы судорог. Если патологий не выявлено, в рацион ребенка вводят белковую пищу, и симптоматика начинается нарастать. Череп такого ребенка несколько уменьшен в размерах. Дети, страдающие патологией, начинают ходить позже сверстников. В год такие пациенты не могут выразить голосом эмоции. Они не воспринимают речь взрослых. Возможна задержка роста.
Не преобразованный фенилаланин выходит с потом и мочой, что приводит к появлению специфического затхлого запаха. Патология проявляется также в своеобразных позах и походке. Они возникают из-за того, что мышечный тонус больного повышен. В положении стоя ребенок широко расставляет ноги и сгибает их в коленях и тазобедренных суставах. При этом голова и плечи опущены. Походка шатающаяся. Ребёнок делает мелкие шаги. Больные сидят в так называемой позе портного. Они поджимают и скрещивают ноги. После 3 лет наблюдается повышенная возбудимость ребенка и его быстрая утомляемость. Присутствует нарушение поведения и психические расстройства. Наблюдается умственная отсталость. Очень часто болезнь сопровождает экзема, дерматит и аритмия. Если лечение отсутствует, состояние больного ухудшится. Своевременная постановка диагноза позволит уменьшить нарушения, с которыми может столкнуться ребёнок.
Описание патологии
Развитие фенилкетонурии (ФКУ) связано с неспособностью организма человека с дефектным геном расщеплять поступающую вместе с пищей, богатой на белки, аминокислоту под названием фенилаланин.
В итоге кислота и элементы ее распада скапливаются в тканях и биожидкостях, превращаются в токсические соединения, под воздействием которых происходит глубокое и в большинстве случаев необратимое поражение органов ЦНС.
Отсутствие своевременного адекватного лечения становится причиной задержки развития интеллекта вплоть до олигофрении.
Манифестирует Фенилкетонурия обычно у больных малышей в первые 3-6 месяцев после рождения.
Выявление заболевания до появления первых клинических проявлений и соблюдение в дальнейшем эллиминационной диеты исключает развитие тяжелых, необратимых расстройств нервной системы у детей и взрослых, то есть опасности для физического, психоэмоционального и умственного здоровья нет.
Фенилкетонурия в среднем диагностируется у 1 новорожденного из 10 тысяч. В Турции количество больных большое – детей с ФКУ рождается 1 на 2, 5 тысячи, в Японии – 1 на 100 тысяч рожденных, в Африке (у представителей негроидной расы) болезнь встречается исключительно редко. У девочек патология диагностируется в 2 раза чаще по сравнению с мальчиками.
В медицинской литературе можно встретить и другие обозначения болезни – фенилпировиноградная олигофрения, болезнь Феллинга.
Симптомы Фенилкетонурии (ФКУ) у детей:
Новорожденный ребенок не похож на больного. Симптомы фенилкетонурии (ФКУ) начинают быть заметны в возрасте 2-6 месяцев. Типичные проявления:
- отсутствие интереса к окружающему миру
- выраженная вялость
- рвота
- беспокойство
- повышенная раздражительность
С 6 месяцев у малыше заметно отставание в психическом развитии. У меньшинства детей это олигофрения в слабой степени. А более чем у половины детей фиксируют идитию. Рост малыша с ФКУ может быть нормальным или сниженным. Зубки режутся поздно, череп может иметь размеры меньше нормы. Сидеть и ходить ребенок с фенилкетонурией начинает поздно.
Детей с рассматриваемым диагнозом можно отличить по позе и походке. Они широко расставляют ноги, сгибая их в тазобедренном и коленных суставах. Шаги мелкие. При ходьбе ребенок покачивается. Сидят они в так называемом положении портного – поджав ноги, поскольку у них повышен мышечный тонус.
При фенилкетонурии (ФКУ) дети обычно имеют голубой цвет глаз и светлый оттенок волос. Кожа почти не пигментирована. От ребенка слышен «мышиный» запах. В некоторых случаях у больного могут быть припадки эпилепсии, но они проходят по мере взросления ребенка.
Другие типичные симптомы ФКУ у детей:
- дермографизм
- потливость
- повышенная чувствительность к солнечным лучам и травмам
- акроцианоз
- тяжёлая экзема
- дерматит
- склонность к запорам
- артериальная гипотония
- расстройства аутистического спектра
- гиперактивность
Если не провести вовремя лечение, уровень интеллекта ребенка будет составлять менее 50. В возрасте 18 месяцев могут появиться судорожные приступы. Исчезают они спонтанно. В раннем возрасте приступы часто проходят в форме инфантильных спазмов, далее становятся тоникоклоническими припадками.
Фенилкетонурия и беременность
Фенилкетонурия, иначе болезнь Феллинга, — это генетическая патология, которая характеризуется нарушением обмена аминокислот
В основе заболевания лежит врожденное изменение метаболизма жизненно важного фермента, определяющего способность и степень умственной деятельности, — фенилаланина. Это важнейшая аминокислота необходима организму для построения молекулы белка
Ее недостаточное образование способствует снижению выработки важнейших биологически активных веществ. Дефицит серотонина и дофамина приводит к отставанию в умственном развитии ребенка, прогрессирующему слабоумию, расстройству мышечного тонуса и психомоторного развития.
Последствия и прогноз жизни
Воздействие излишнего количества фенилаланина на нервную систему ребенка приводят к стойким психологическим нарушениям. Уже к 4 годам, без должного лечения, больные фенилкетонурией дети причисляются к слабоумным и физически недоразвитым членам общества. Они пополняют ряды инвалидов детства и краски жизни меркнут для них.
Не искрится счастьем и жизнь родителей больного ребенка. Малыш требует постоянного ухода, и при ограниченных финансовых средствах это выливается в общее ухудшение благосостояния семьи. Боль, испытываемая мамой и папой от невозможности изменить существование ребенка в лучшую сторону угнетает и давит, но отчаиваться нельзя. Помогите себе, помогите ребенку пройти эти испытания с меньшими потерями в любви и милосердии.
Наука спешит, она делает семимильные шаги в направлении исключения заболевания из ранга тяжелых. Огромное значение имеет диагностика фенилкетонурии в утробе матери, но пока такого метода не изобретено. «пока» не значит «никогда», будем ждать и верить
Лечение
Лечение заключается в резком ограничении поступающего с пищей белка (в зависимости от толерантности к нему организма), а следовательно, и фенилаланина. Количество последнего не должно превышать 15—40 мг/кг (в первые месяцы жизни — 50—60 мг/кг). Из пищи исключают продукты с высоким содержанием белка: мясо, рыбу, яйца, сыр, молоко, горох, фасоль и др. Разрешают сахар, фруктовые соки, натуральный и искусственный мед, растительные масла, искусственное саго. В качестве заменителей белка используют различные препараты, в к-рых полностью или почти полностью отсутствует фенилаланин (лофеналак, цимогран, берлофен, минафен). Молоко, картофель, хлеб, овощи и фрукты назначают только после тщательного подсчета содержания в них фенилаланина. Диету следует соблюдать не менее 6—7 лет, а по мнению некоторых исследователей, и дольше. Диета необходима также больным фенилкетонурией женщинам во время беременности для избежания вредного воздействия кетокислот, накапливающихся в организме женщины, на плод.
Особенности питания новорожденных и диетотерапия
На первом году жизни ребенка с ФКУ допустимо кормление грудным молоком, но его количество должно быть ограничено. До 6 месяцев допустимым уровнем потребления фенилаланина является 60-90 мг на 1 кг веса малыша (в 100 г молока содержится 5,6 мг фенилаланина). Начиная с 3 месяцев, рацион ребенка следует поэтапно расширять, вводя в него фруктовые соки и пюре.
Детям с 6 месяцев разрешено введение в рацион овощных пюре, каш (из саго), безбелковых киселей. После 7 месяцев можно давать малышу низкобелковые макаронные изделия, с 8 месяцев – хлеб, не содержащий белка. Возраст, до которого следует ограничивать поступление белка в организм больного ребенка, не установлен. Врачи до настоящего времени дискутируют по вопросу целесообразности пожизненной диетотерапии, но сходятся во мнении, что минимум до 18 лет необходимо придерживаться диетического питания.
Фенилкетонурия, диагностированная у женщины, не является поводом отказаться от рождения ребенка. Будущим мамам с ФКУ для предупреждения повреждения плода во время беременности и профилактики возможных осложнений необходимо до планируемого зачатия и во время вынашивания ребенка придерживаться диеты с ограничением фенилаланина (его уровень в крови должен быть до 242 мкмоль/л).
Безлактозные смеси для малышей
Диета при фенилкетонурии базируется на существенном уменьшении дозы натурального белка в суточном рационе, но организм новорожденного ребенка не может развиваться нормально при отсутствии необходимых микроэлементов. Для восполнения потребности малыша в белке применяются безлактозные аминокислотные смеси, которыми, согласно российскому законодательству, больные должны обеспечиваться бесплатно.
Толерантность грудничка к фенилаланину в течение первого года жизни стремительно изменяется, поэтому необходимо контролировать его концентрацию в крови ребенка и вносить корректировки в диету. Смеси рассчитаны на определенные возрастные группы:
- малышам до года назначаются Афенилак 15, Аналог-СП, PKU-1, PKU-mix, PKU Anamix;
- детям, старше 1 года, назначают обогащенные витаминами и минералами смеси с повышенным содержанием белка – PKU Prima, P-AM Universal, ФКУ-1, ФКУ-2, ХР Максамейд, ХР Максамум.

Диетические продукты для пополнения запасов белка
Одним из основных компонентов пищевой диеты при фенилкетонурии являются малобелковые продукты на основе крахмала. Эти добавки содержат гидролизат казеина, триптофан, тирозин, метионин, азот и обеспечивают суточную потребность организма ребенка в белке, который необходим для нормального развития и роста. Специализированными продуктами, восполняющими нехватку необходимых минералов и аминокислот при их нехватке в рационе питания, являются:
- Берлофен;
- Циморган;
- Минафен;
- Апонти.
Беременность и ФКУ
Если беременность наступает у женщин с ФКУ, не получающей адекватного диетического лечения, развивающийся плод повреждается.
У новорожденного выявляются неврологические симптомы и врожденные пороки.
Высокие уровни фенилаланина токсично действуют на мозг плода.
Хотя ребенок и не болеет фенилкетонурией, но он повреждается аномальными метаболитами матери
Это явление получило название «материнская фенилкетонурия»
Более 90% детей, рожденными матерями с нелеченной ФКУ, имеют умственную отсталость, микроцефалию, пороки сердца, задержку внутриутробного развития плода.
Диетическое лечение таких детей бесполезно, так как они не страдают фенилкетонурией
Профилактика материнской ФКУ– это наша ответственность
-
ВОПРОСЫ:
-
Существуют методы лечения?
-
Каковы прогнозы?
-
Как предотвратить эту катастрофу?
-
ОТВЕТЫ:
-
Учитывая значительную степень повреждения плодов женщин с гиперфенилаланинемией профилактически должны быть применены:
-
ригористическая диета с низким содержанием ФА
-
планирование семьи
-
РИГОРИСТИЧЕСКАЯ диета у беременной женщины — начать надо раньше, трудно переводить взрослую женщину на диету, которая по своей строгости напоминает таковую у младенцев
-
Молодые женщины, способные к деторождению, должны постоянно соблюдать диету
-
Строгое соблюдение диеты как минимум 3 месяца до зачатия
-
Переход на ригористическую диету в 1 триместре вызывает больший дискомфорт, чем плавная смена диеты до зачатия
-
„обыкновенные” проблемы больного человека значительно обостряются
Симптомы заболевания
При своевременном обнаружении болезнь Феллинга поддается успешному лечению путем корректировки питания, и развитие ребенка происходит в соответствии его возрастной группе. Трудность выявления генной мутации заключается в том, что ранние признаки тяжело обнаружить даже опытному педиатру. Выраженность симптоматики врожденного заболевания усиливается по мере взросления ребенка, потому что употребление белковой пищи способствует развитию нарушений ЦНС.
Признаки у новорожденных
На протяжении первых дней жизни ребенка признаки патологических отклонений обнаружить трудно – малыш ведет себя естественно, задержки в развитии не наблюдается. Симптомы заболевания впервые начинают проявляться через 2-6 месяцев после рождения. Родителей должно насторожить поведение малыша, которое характеризуется низкой активностью, вялостью, или, наоборот, беспокойством, гипервозбудимостью.
С началом грудного вскармливания в организм новорожденного с молоком начинают поступать белки, что служит катализатором появления первых признаков, однозначно свидетельствующих о том, что заболевание начало прогрессировать. К специфическим клиническим проявлениям болезни относятся:
- постоянная рвота (зачастую принимаемая за врожденное сужение привратника);
- частое срыгивание;
- отсутствие реакции на внешние раздражители;
- мышечная дистония (сниженное напряжение мышц);
- судорожный синдром (судороги эпилептического или неэпилептического характера).
Симптомы у детей после 6 месяцев
Если манифестация генетической болезни не произошла (или не была замечена) в течение первых 6 месяцев с момента рождения ребенка, то после этого периода уже можно точно определить отставание в психомоторном развитии. Симптомами генетических нарушений, вызванных ферментным дефицитом, у детей, старше полугода, являются:
- снижение активности (вплоть до полной безучастности);
- отсутствие попыток к самостоятельному вставанию, сидению;
- особенный «мышиный» запах кожи (запах плесени возникает вследствие выведения токсических производных фенилаланина через потовые железы и мочу);
- потеря способности к визуальному распознаванию лиц родителей;
- шелушение кожи;
- появление дерматитов, экзем, склеродермии.
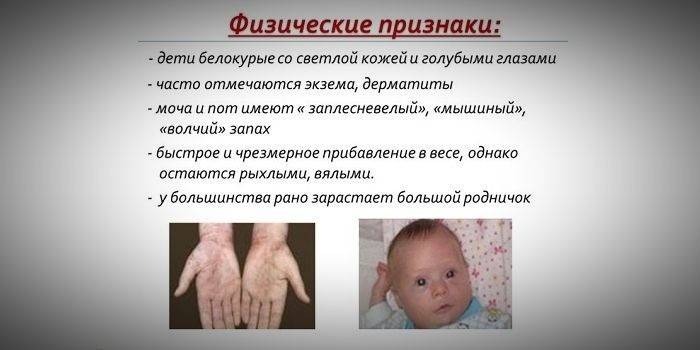
Прогрессирование заболевания при отсутствии лечения в детском возрасте
Если отклонения в развитии не были выявлены в младенческом возрасте, и соответствующее лечение не проводилось, то заболевание начинает активно прогрессировать и нередко приводит к инвалидности. Отсутствие терапии на раннем этапе болезни вызывает появление следующих симптомов болезни уже в возрасте 1,5 лет:
- микроцефалия (уменьшенные размеры головного мозга);
- прогнатия (смещение верхнего зубного ряда вперед);
- позднее прорезывание зубов;
- гипоплазия эмали (истончение или полное отсутствие зубной эмали);
- задержка речевого развития вплоть до полного отсутствия речи;
- 3, 4 степень олигофрении (задержка психического развития, умственная отсталость);
- врожденные пороки сердца (дефекты в структуре сердечной мышцы, отделах сердца, крупных сосудах);
- расстройства вегетативной системы (акроцианоз, повышенная потливость, артериальная гипотония);
- запоры.
Причины фенилкетонурии

В основе заболевания лежит генетический дефект – мутация гена 12-й хромосомы (98% всех случаев фенилкетонурии). Это так называемая классическая фенилкетонурия. Ген кодирует количество фермента фенилаланин-4-гидроксилазы. Фермент отвечает за превращение аминокислоты фенилаланина в организме человека в тирозин в клетках печени. Фенилаланин – это аминокислота, которая содержится в белковых продуктах (мясо, рыба, молоко, яйца и другие).
При мутации гена количество фермента снижается, что приводит к накоплению фенилаланина и продуктов промежуточного метаболизма в тканях организма. Организм пытается избавиться от фенилаланина и продуктов его распада, выводя их с мочой.
Подобные нарушения обмена веществ приводят к нарушению строения нервных проводников, снижению образования нейромедиаторов. Все это, наряду с прямым токсическим действием избытка фенилаланина, приводит к развитию умственных нарушений, являющихся основным проявлением заболевания.
Фенилкетонурия наследуется по аутосомно-рецессивному типу, то есть не зависит от пола, и возникает при совпадении двух патологических генов от отца и матери.
Остальные 2% случаев фенилкетонурии связаны с другими генетическими дефектами и зависят от концентрации прочих ферментов (дигидроптеридинредуктазы и др.). Они имеют те же клинические проявления, но не поддаются лечению диетой. Такие варианты относят к атипичному течению заболевания. Среди них принято выделять фенилкетонурию II и III. Генетический дефект при фенилкетонурии II располагается в 4-й хромосоме, при III – в 11-й хромосоме.
Непростительные ошибки
Педиатр просит сдать анализы, на руках талон, время поджимает, а собрать мочу никак не получается. Знакомая ситуация?
Даже в этом случае не следует совершать таких ошибок:
- собирать материал в течение дня, хранить его до утра, а потом нести в лабораторию. Понятно, что анализ мочи у грудничка будет недостоверным и его придется повторить;
- использовать для сбора образца подручные средства, совершенно для этого не подходящие, например, выжимать жидкость с пеленки или переливать в баночку из тарелки. Этот образец изначально будет не качественным, поскольку в нем волокно, пыль и прочее;
- пусть даже в небольших количествах отпаивать ребенка водой или соком, для новорожденных это совершенно недопустимо. Кормить грудным молоком и смесью чаще обычного для обильного мочеиспускания – это максимум;
- не подмывать ребенка, а сразу приступать к процессу.
Для сбора мочи есть масса вариантов, нужно лишь выбрать тот, что подходит маме и ее дочери, будь то детский мочеприемник или стеклянная банка. Главное — не бояться и не нервничать при первой же неудаче. Со временем, по мере накопления опыта, сделать это будет проще.
Как без проблем собрать мочу на анализ у ребенка
https://youtube.com/watch?v=UE3WR9YkxFE
Судя по количеству людей, мучающихся этой проблемой, производители педиатрических мочеприемников не смогли составить грамотной инструкции к своему продукту. Действительно, число мам и пап, штудирующих интернет накануне предстоящей сдачи анализов у малыша превышает разумное число. И не удивительно. Например, инструкция к мочеприемнику, который купила я, начинается со следующих слов (дословно): “приклейте нижнюю часть клеевой пластины…”
Я испортила несколько штук, пока разобралась где у этой клеевой пластины верх, а где низ. Возможно, вы окажетесь догадливее меня и вам понадобится всего один, но на всякий случай ниже приведена подробная инструкция что и куда надо приклеить для того, чтобы собрать у новорожденного мочу с помощью педиатрического мочеприемника 🙂
Если нет особых инструкций педиатра, для простого общего анализа мочи собирать лучше утреннюю мочу (бывают разные типы анализов, когда, например, собирают суточную мочу).
Количество мочи для анализа
Будет достаточно, если небольшая банка (размером, как банка от детского питания), в которую вы перельете мочу, будет заполнена на сантиметр. Тару нужно предварительно тщательно вымыть с моющим средством и простерилизовать (или хотя бы сполоснуть кипятком).
| 1. Имеем вот такой запечатанный стерильный педиатрический мочеприемник. | |
| 2. Моем руки с мылом. Вскрываем наружный пакет. Достаем стерильный мешочек.Выглядит он так: |  |
| 3. Отрываем бумажку от липкой ленты. |  |
4. Теперь наш мочеприемник готов к тому, чтобы его прилепили.
Положите ребенка на спину, раздвиньте ножки (предварительно тщательно его подмыв). Убедитесь, что промежность сухая.

5. Кликните на картинку, чтобы увеличить ее.
Желтый крестик- тот самый низ клеевой пластины. Он должен оказаться между анусом и половым органом (это делают для того, чтобы каки не попали в анализ мочи).
1. Мальчику- вставляем пенис в отверстие в липкой пластине и плотно прижимаем клеевую часть к коже.
2. Девочке – начинаем клеить с желтого крестика, приклеиваем крестик к участку кожи, расположенному между половыми губами и анальным отверстием. Затем приклеиваем всю липкую полосу, двигаясь от крестика к лобковой зоне.
6. Приклеили и ждем-с 🙂
Ребенка можно аккуратно одеть в свободную одежду или завернуть в одеяло, чтобы не замерз.
Чтобы не испортить диван, подстелите под малыша что-нибудь.
Для убыстрения процесса – покормите грудью, дайте попить водички, включите кран с водой.
Хороших вам анализов!
Помогла статья? Нажмите лайк или оставьте комментарий на сайте:
Online-консультации врачей
| Консультация аллерголога |
| Консультация общих вопросов |
| Консультация специалиста по лазерной косметологии |
| Консультация онколога-маммолога |
| Консультация пульмонолога |
| Консультация трихолога (лечение волос и кожи головы) |
| Консультация детского психолога |
| Консультация ортопеда-травматолога |
| Консультация эндокринолога |
| Консультация радиолога (диагностика МРТ, КТ) |
| Консультация проктолога |
| Консультация вертебролога |
| Консультация эндоскописта |
| Консультация гастроэнтеролога |
| Консультация гомеопата |
Новости медицины
Футбольные фанаты находятся в смертельной опасности,
31.01.2020
«Умная перчатка» возвращает силу хвата жертвам травм и инсультов,
28.01.2020
Назван легкий способ укрепить здоровье,
20.01.2020
Топ-5 салонов массажа в Киеве по версии Покупон,
15.01.2020
Новости здравоохранения
Глава ВОЗ объявил пандемию COVID-19,
12.03.2020
Коронавирус атаковал уже более 100 стран, заразились почти 120 000 человек,
11.03.2020
Коронавирус атаковал 79 стран, число жертв приближается к 3200 человек,
04.03.2020
Новый коронавирус атаковал 48 стран мира, число жертв растет,
27.02.2020
Скрининг
Кровь берется у двухнедельного ребенка на фенилкетонурию.
Во многих странах ФКУ обычно включается в группы по скринингу новорожденных с использованием различных методов выявления. Большинство детей в развитых странах проходят скрининг на ФКУ вскоре после рождения. Скрининг на ФКУ проводится с помощью анализа ингибирования бактерий ( тест Гатри ), иммуноанализа с использованием флуорометрического или фотометрического определения или измерения аминокислот с использованием тандемной масс-спектрометрии (МС / МС). Измерения, проведенные с помощью MS / MS, определяют концентрацию Phe и отношение Phe к тирозину , это соотношение будет повышено в PKU.
Патогенез
При фенилкетонурии в печени больного не продуцируется особый фермент под названием фенилаланин-4-гидроксилаза.
Основная его функция – преобразование поступающего в органы ЖКТ с пищей фенилаланина в аминокислоту тирозин. Это аминокислота входит в состав большинства ферментов, гормонов, способствует выработке меланина (пигмент), принимает участие в образовании белков и в функционировании большинства внутренних органов.

Метаболический блок при фенилкетонурии неуклонно приводит к тому, что начинают работать побочные (атипичные) пути обмена фенилаланина, в результате он трансформируется в те вещества, которых в теле здорового человека быть не должно.
Это кислоты – фенилмолочная и фенилпировиноградная, ортофенилацетат, фенилэтиламин. По сути, они являются токсическими соединениями, и их накапливание в кровеносной системе приводят:
- К нарушению нормального обмена липидов (жировых клеток) в разных отделах головного мозга;
- К нехватке нейромедиаторов, ответственных за бесперебойную передачу нервных импульсов во всей нервной системе.
В результате это приводит к прогрессирующему снижению интеллекта и к умственной отсталости выраженной степени – олигофрении, имбецильности, идиотии.
Отсутствие терапии становится причиной того, что через каждые 10 недель коэффициент развития интеллекта больного малыша падает на 5 пунктов.
Наследование
Так как фенилкетонурия наследуется как рецессивный признак, то для проявления его у ребенка необходимо, чтобы у обоих родителей имелся дефектный ген. Именно поэтому родственные браки во многих странах запрещены.
Если рассматривать случай рождения детей в обычной семье, то у носителей такой мутации они могут быть:
- 25% вероятности, что ребенок родится больным.
- В 50% случаев малыш здоров, но является носителем дефектного гена.
- Четвертая часть потомства будет абсолютно нормальной.

Эта схема не дает полной картины рождаемости больных детей. Она только отражает вероятность, поэтому у каждой семейной пары процент дефектных генов может быть свой, и предсказать исход, к сожалению, невозможно. Сейчас существуют консультации, в которых специалисты-генетики помогают парам спрогнозировать рождение у них больного ребенка, рассказывая при этом, как фенилкетонурия наследуется.
Противоречия в законе
По словам Слепцовой, она неоднократно пыталась добиться от Минздрава бесплатного предоставления низкобелковых продуктов в соответствии с 890-м постановлением. Однако чиновники ей отказали (копия ответа есть в распоряжении RT).
Также по теме

«Первый список пациентов сформирован»: глава «Круга добра» о работе фонда, лечении СМА и действиях чиновников в регионах
Фонд помощи тяжелобольным детям «Круг добра» уже сформировал первый список пациентов, в том числе со спинальной мышечной атрофией, для…
«Пришёл ответ, где они ссылаются на приказ Минздрава №737н о стандартах специализированной медпомощи детям при классической ФКУ (такая форма болезни наиболее распространена. — RT) и говорят, что там прописана только низкобелковая диета и специализированные продукты лечебного питания (смеси). А безбелковых продуктов там не указано, соответственно, они нам ничего не должны. Но приказ Минздрава не может противоречить постановлению правительства, он ниже по субординации», — подчёркивает Татьяна.
В ответе ведомства сообщается, что в ФЗ №323 «Об основах охраны здоровья» в ст. 39 п. 3 прописано понятие специализированных продуктов лечебного питания, продолжает Слепцова. «Но в этой же статье в п. 4 прописано, что нормы этого питания утверждаются уполномоченным федеральным органом исполнительной власти. Так вот, приказом Минздрава «Об утверждении норм лечебного питания» (№395н от 21.06.2013) утверждена таблица с разными диетами — стандартной, щадящей, низкокалорийной, низкобелковой и так далее. Там расписано, сколько граммов в сутки каких продуктов человеку положено при каждой из них. И в этих нормах прописано, что при низкобелковой диете, которую предписывает приказ Минздрава о стандарте медпомощи при ФКУ, в рационе есть мясо, крупы, бобовые, четверть яйца в сутки», — объясняет она.
В то же время в клинических рекомендациях по лечению ФКУ, выпущенных Минздравом, говорится, что из рациона больного исключают мясо, мясопродукты, рыбу, рыбопродукты, творог, яйца, бобовые, орехи и друге продукты.